

批量获取NCBI序列
概念介绍
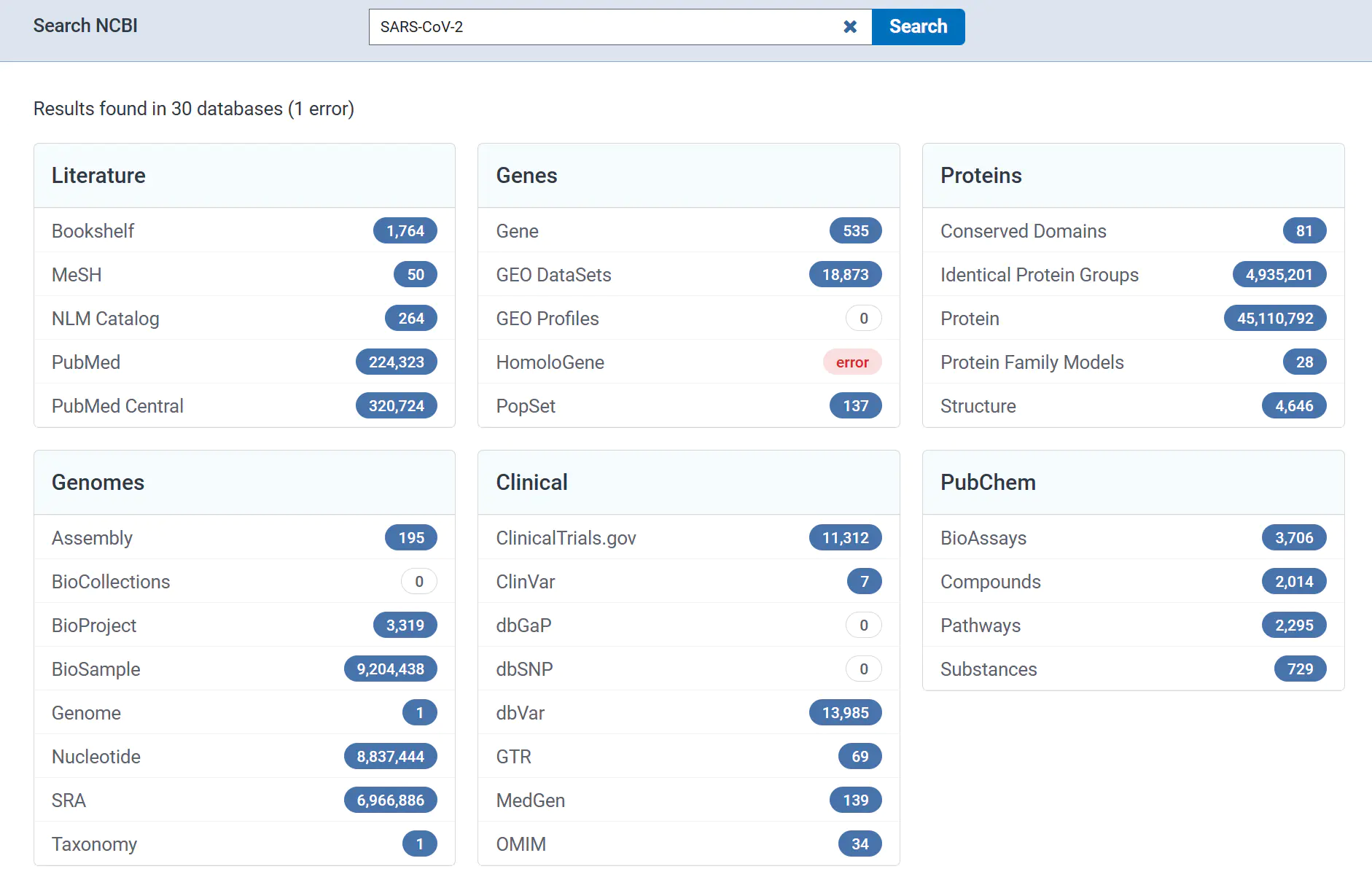
NCBI(National Center for Biotechnology Information)是美国国立卫生研究院(NIH)下属的机构,在其官网上提供了统一搜索引擎,可以查询多种数据库
最常用的数据库包括
- PubMed:检索文献,定位类似Google Scholar、Web of Science
- nucleotide:核酸序列,包括基因组、基因、转录本等。由GenBank及其他数据库提供支持
- SRA:原始测序reads
- GEO:上游分析后得到的基因表达矩阵
- dbSNP:单核酸多态性数据
Entrez是NCBI开发的一个集成搜索工具,目前有命令行版(edirect)、Python版(Biopython.Entrez)等
本文将使用Python从nucleotide数据库批量获取序列信息
下载序列
主要用到 Biopython包的 Entrez模块。需要设置一个email地址
from Bio import Entrez
from Bio import SeqIO
Entrez.email = "your.email@example.com"
使用 Entrez.efetch函数获取数据,db指定数据库为 nucleotide。要求返回的类型是 gb,即GenBank类型,格式为 text纯文本,方便Biopython读取
def fetch_sequence(id):
with Entrez.efetch(db="nucleotide", rettype="gb", retmode="text", id=id) as handle:
record = SeqIO.read(handle, "genbank")
return record
seq = fetch_sequence("OQ350818.1")
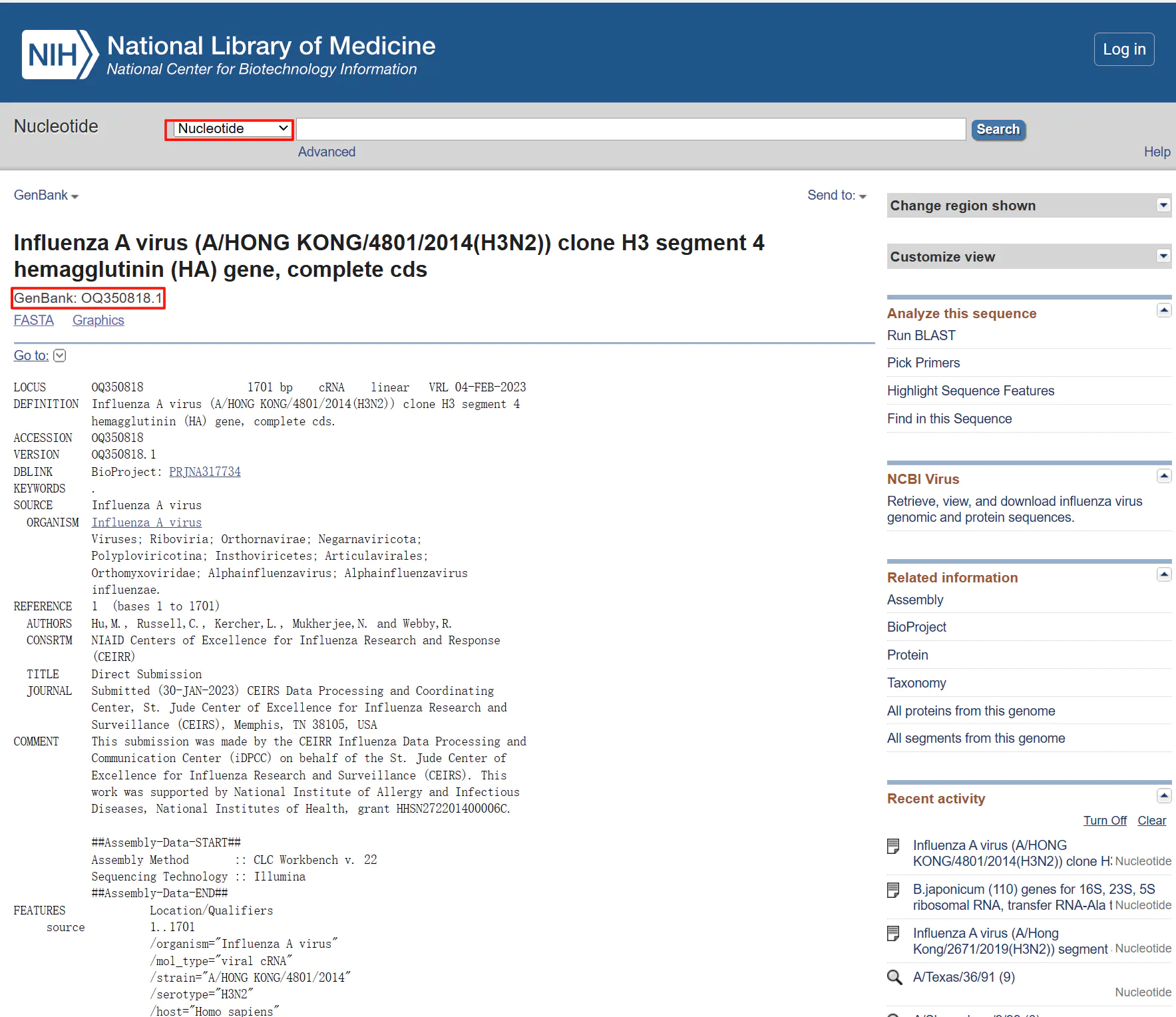
seq是 Bio.SeqRecord.SeqRecord对象,如下
SeqRecord(seq=Seq('ATGAAGACTATCATTGCTTTGAGCTACATTCTATGTCTGGTTTTCGCTCAAAAA...TGA'), id='OQ350818.1', name='OQ350818', description='Influenza A virus (A/HONG KONG/4801/2014(H3N2)) clone H3 segment 4 hemagglutinin (HA) gene, complete cds', dbxrefs=['BioProject:PRJNA317734'])
使用 dir(seq),可列出该对象所有的方法、属性。其中
id:唯一iddescription:基本描述seq:核酸序列features:特征,包括CDS的注释、翻译后蛋白序列annotations:注释信息,包括分子类型(molecule_type)、物种分类(taxonomy)、提交日期(date)等dbxrefs:其他数据库(如BioProject)的链接id
我们的目的即通过 seq.seq获取核酸序列,通过 seq.features获取蛋白序列
nt = seq.seq
cds = [ ft for ft in seq.features if ft.type == 'CDS'][0]
aa = cds.qualifiers['translation'][0]
注意如果该序列对应了多个CDS,以上代码只能获取第一个CDS feature的蛋白序列
至此,我们实现了从id获取核酸、蛋白序列,写个循环即可实现批量获取
检索id
以上,我们假定你已经有了id列表,然后可以循环下载序列。但其实前期寻找id也可以借助代码完成,比在浏览器中操作快得多
我们使用 Entrez.esearch,可以检索关键词,并获取检索结果的id列表。每个条目有accession id(GenBank id)、GI id两种唯一id,前者更为通用。当前版本(v1.81)的Biopython默认返回GI id列表,但我们最终通过 Entrez.esummary获取accession id,因此对结果无影响
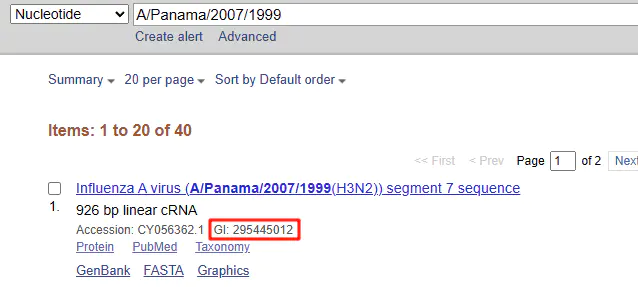
获取id列表后,就可以使用 Entrez.esummary获取每个条目的基本信息,并对基本信息做些筛选。在本案例中,对序列长度、标题关键词做了筛选
通过筛选的条目,就可以打印出accession id、长度、标题,方便我们确认
def search_summary(query, max_results=10, min_length=100, keyword=None):
with Entrez.esearch(db="nucleotide", term=query, retmax=max_results) as handle:
record = Entrez.read(handle)
id_list = record["IdList"]
with Entrez.esummary(db="nucleotide", id=",".join(id_list)) as handle:
summary_records = Entrez.read(handle)
for summary in summary_records:
if int(summary['Length']) < min_length:
continue
if keyword:
if keyword not in summary['Title']:
continue
print(f"ID: {summary['AccessionVersion']}, Length: {int(summary['Length'])}")
print(f"Title: {summary['Title']}")
search_summary('A/Panama/2007/1999', keyword='HA', min_length=1700)
输出如下
ID: KM821317.1, Length: 1739
Title: Influenza A virus (A/Panama/2007/1999(H3N2)) segment 4 hemagglutinin (HA) gene, complete cds
代理
有时直接访问NCBI网络连接不稳定,可通过代理访问。由于 Bio.Entrez底层的请求库是 urllib,我们为 urllib设置默认的代理服务器即可
import urllib.request
def set_global_proxy(proxy_address):
proxy_config = { 'http': proxy_address, 'https': proxy_address}
proxy_handler = urllib.request.ProxyHandler(proxy_config)
opener = urllib.request.build_opener(proxy_handler)
urllib.request.install_opener(opener)
proxy = "http://your_proxy_url:7890" # 替换为自己的代理服务器
set_global_proxy(proxy)
设置后,可以访问测试网址,看能否返回html
target_url = 'https://www.google.com'
response = urllib.request.urlopen(target_url)
html = response.read().decode('utf-8')
print(html)
如果设置成功,会返回 <!doctype html>开头的html文本。此时再进行检索、下载操作,就稳定得多